新版TCGA数据库学习:提取新版TCGA表达矩阵(tpm/count/fpkm) |
您所在的位置:网站首页 › stranded deep指令 › 新版TCGA数据库学习:提取新版TCGA表达矩阵(tpm/count/fpkm) |
新版TCGA数据库学习:提取新版TCGA表达矩阵(tpm/count/fpkm)
|
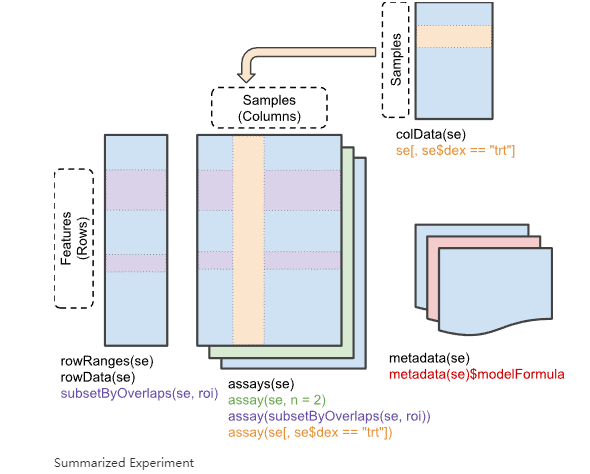
现在使用TCGAbiolinks下载转录组数据后,直接是一个SummarizedExperiment对象,这个对象非常重要且好用。因为里面直接包含了表达矩阵、样本信息、基因信息,可以非常方便的通过内置函数直接提取想要的数据,再也不用手扒了!! 这个对象的结构是这样的: 是不是感觉和单细胞的SingCellExperiment对象非常像~
上次我们下载了常见的组学数据,今天学习下怎么提取数据,就以TCGA-READ的转录组数据为例。 分别提取mRNA和lncRNA的表达矩阵,还要添加gene symbol的那种! 加载数据和R包加载之前下载好的数据。 rm(list = ls()) library(SummarizedExperiment) ## Loading required package: MatrixGenerics ## Loading required package: matrixStats ## ## Attaching package: 'MatrixGenerics' ## The following objects are masked from 'package:matrixStats': ## ## colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse, ## colCounts, colCummaxs, colCummins, colCumprods, colCumsums, ## colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs, ## colMads, colMaxs, colMeans2, colMedians, colMins, colOrderStats, ## colProds, colQuantiles, colRanges, colRanks, colSdDiffs, colSds, ## colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads, ## colWeightedMeans, colWeightedMedians, colWeightedSds, ## colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet, ## rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods, ## rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps, ## rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins, ## rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks, ## rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars, ## rowWeightedMads, rowWeightedMeans, rowWeightedMedians, ## rowWeightedSds, rowWeightedVars ## Loading required package: GenomicRanges ## Loading required package: stats4 ## Loading required package: BiocGenerics ## ## Attaching package: 'BiocGenerics' ## The following objects are masked from 'package:stats': ## ## IQR, mad, sd, var, xtabs ## The following objects are masked from 'package:base': ## ## anyDuplicated, append, as.data.frame, basename, cbind, colnames, ## dirname, do.call, duplicated, eval, evalq, Filter, Find, get, grep, ## grepl, intersect, is.unsorted, lapply, Map, mapply, match, mget, ## order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank, ## rbind, Reduce, rownames, sapply, setdiff, sort, table, tapply, ## union, unique, unsplit, which.max, which.min ## Loading required package: S4Vectors ## ## Attaching package: 'S4Vectors' ## The following objects are masked from 'package:base': ## ## expand.grid, I, unname ## Loading required package: IRanges ## ## Attaching package: 'IRanges' ## The following object is masked from 'package:grDevices': ## ## windows ## Loading required package: GenomeInfoDb ## Loading required package: Biobase ## Welcome to Bioconductor ## ## Vignettes contain introductory material; view with ## 'browseVignettes()'. To cite Bioconductor, see ## 'citation("Biobase")', and for packages 'citation("pkgname")'. ## ## Attaching package: 'Biobase' ## The following object is masked from 'package:MatrixGenerics': ## ## rowMedians ## The following objects are masked from 'package:matrixStats': ## ## anyMissing, rowMedians load("TCGA-mRNA/TCGA-READ_mRNA.Rdata") se |
【本文地址】
今日新闻 |
推荐新闻 |